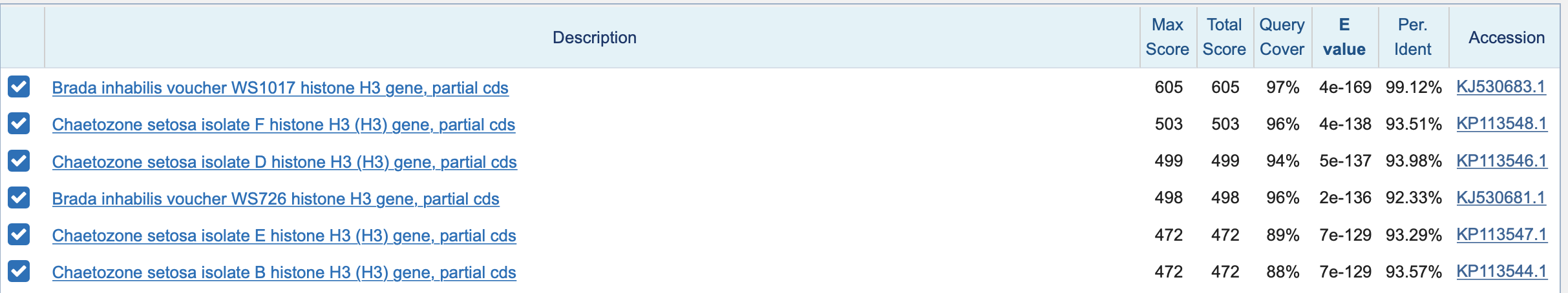
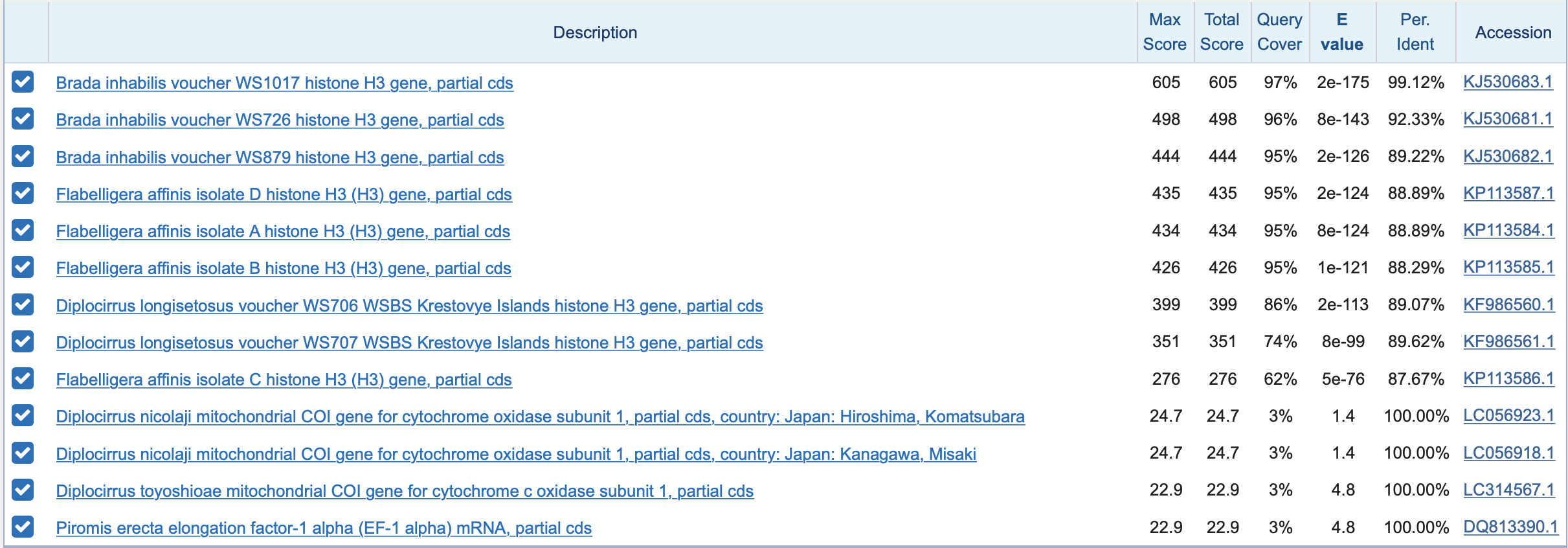
Для выполнения задания была использована консенсусная последовательность из практикума 6. Чтобы найти похожие последовательности(см.картинку) я воспользовалась BLASTn.
Для определения таксономии я перешла в раздел Taxonomy и посмотрела на предложенные варианты. Заметим, что самый большой показатель Score имеет вид Brada. А также у Brada inhabilis наибольший %identity.
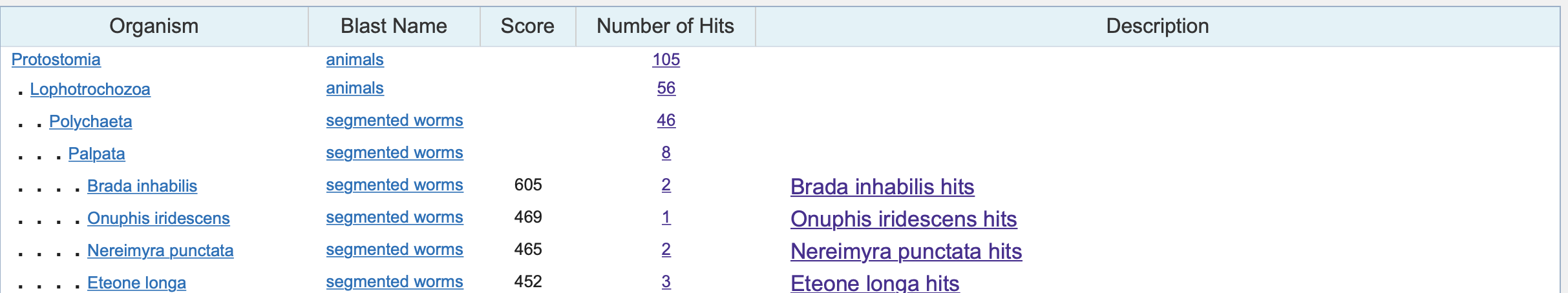
Судя по выравниванию, может определить, что последовательность аналогична(за исключением неопределнных хроматограммой нуклеотдов) Brada inhabilis.
Воспользовавшись BLASTx, можно сделать вывод, что последовательность содержит ген гистон H3.

В этом задании требуется сравнить megablast, blastn с параметрами по умолчанию и blastn с самыми чувствительными настройками. Чувствительные настройки-word size и mismatch score. В качестве ограничения поиска я выбрала семейство Flabelligerida.
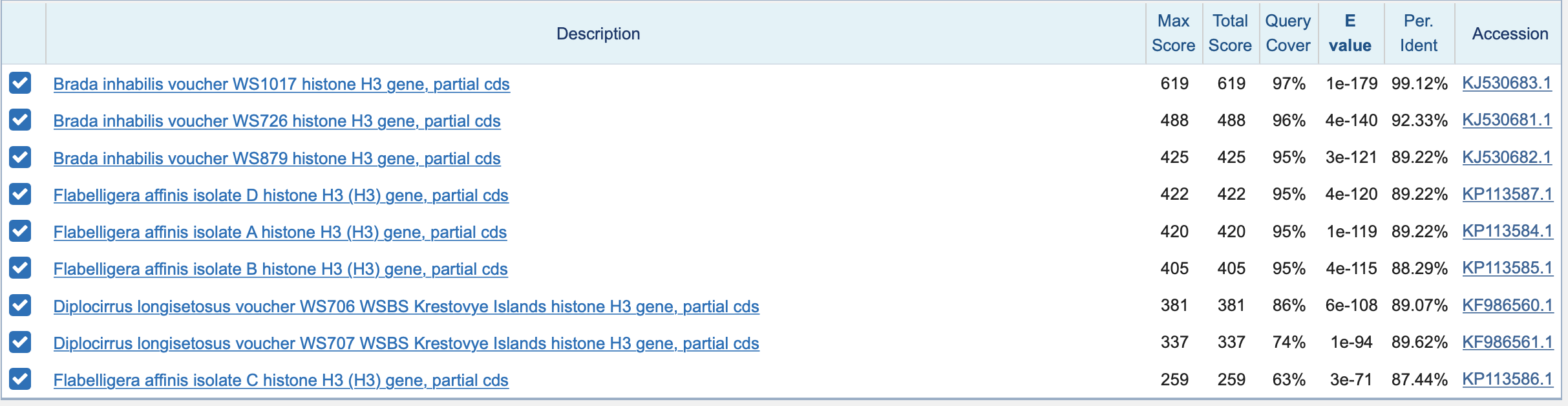
1)При запуске измененного blastn получается 22 находки. Среди них есть и те, у которых E-value>>0.001 (например равное 7.2)
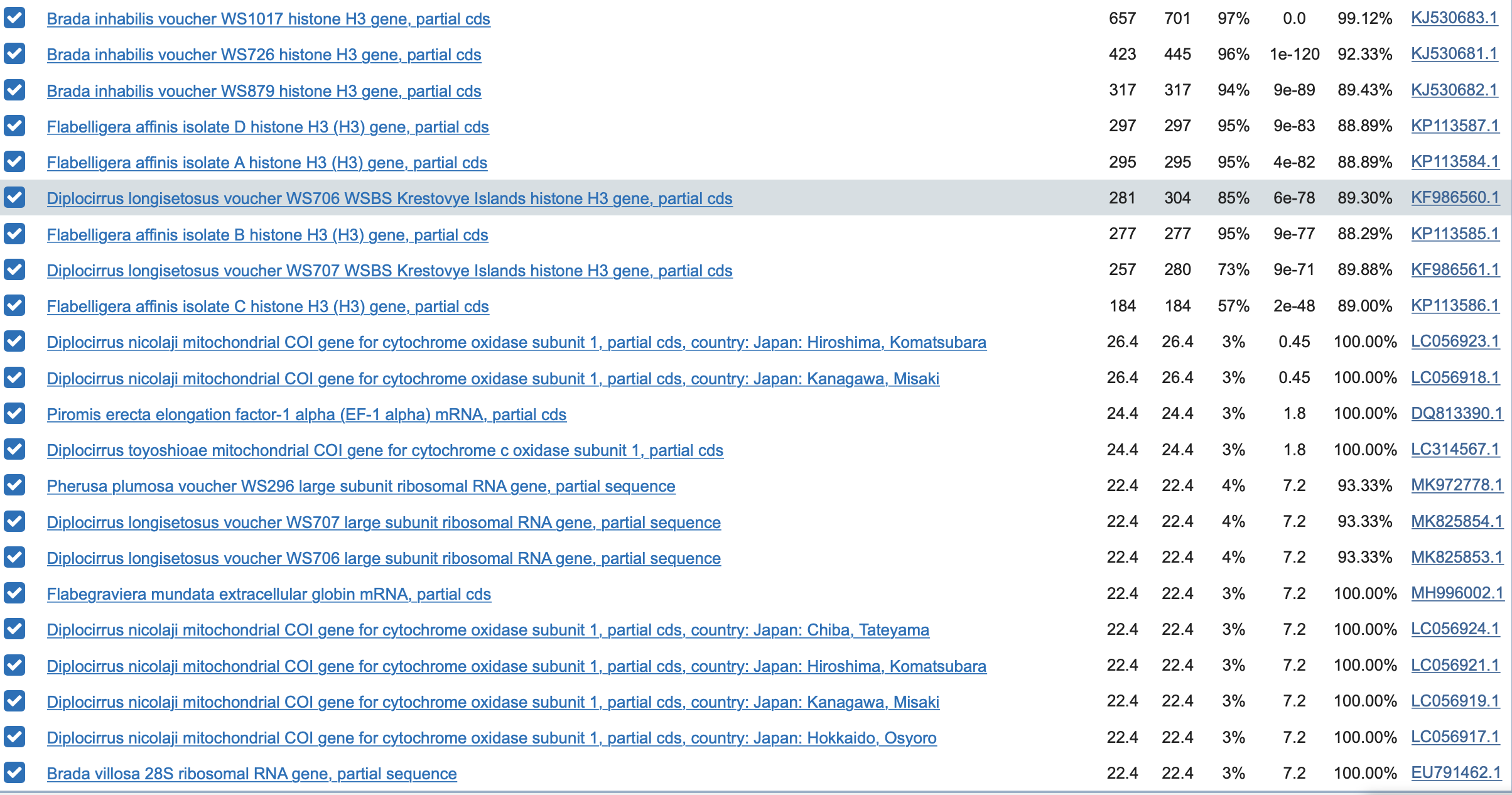
2) Blastn выдает 13 находок, причем ни у одной теперь нет E-value=0.0 и максимальное значение снизилось хотя бы до 4.8.
3) В megablast 9 находок, но с меньшим значением E-value чем в blastn.
| Алгоритм | Параметры | Число находок |
| megablast | Автоматические Длина слова = 28; Match/Mismatch scores 1, -2 | 9 |
| blastn | Автоматические Длина слова = 11; Match/Mismatch scores 2, -3 | 13 |
| blastn с изменениями | Изменённые Длина слова = 7; Match/Mismatch scores 1, -4 | 22 |
Таким образом можно сделать вывод, что что megablast выдаёт более сходные последовательности, чем blastn, что можно сказать и про blastn чувствительный(видим e-value=0), но в отличие от него не выдает последовательности с высокими значениями E-value, что означает отсутсвтие недостоверных находок megablast. Плюс еще megablast работает быстрее других.
Теперь тоже самое проделаю с CDS вируса Microbacterium phage TeddyBoy из практикума 7.
Blastn обычный выдает много находок (155), но больше половины из них недостоверны(e-value>>0.001) (поиск ограничила семейством Siphoviridae)

Blastn с измененными параметрами выдает 90 находок, появились последовательности с e-value=0.0, но число недостоверных все еще примерно половина.
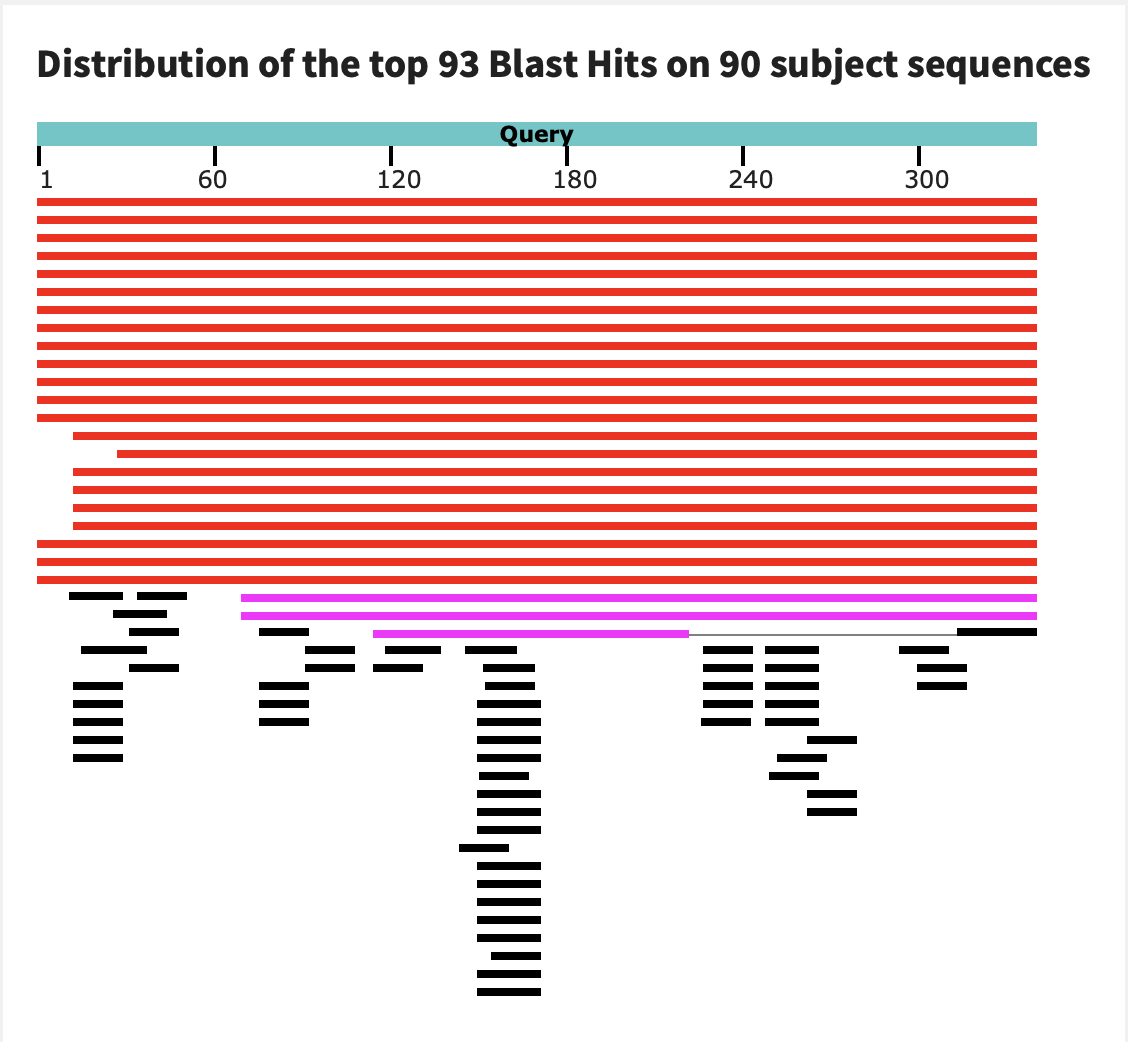
Megablast выдал 24 находки. Нет e-value>>0.001, т.е. все находки достоверные. Снова делаем вывод, что megablast самый удобный для поиска достоверных находок
| Алгоритм | Параметры | Число находок |
| megablast | Автоматические Длина слова = 28; Match/Mismatch scores 1, -2 | 24 |
| blastn | Автоматические Длина слова = 11; Match/Mismatch scores 2, -3 | 155 |
| blastn с изменениями | Изменённые Длина слова = 7; Match/Mismatch scores 1, -4 | 90 |
Теперь нужно проверить есть ли гомологи белков в геноме организма Amoboaphelidium protococarum (сборка генома в файле на kodomo /P/y18/term3/block2/X5.fasta). Для этого я выбрала три белка, которые точно есть у эукариот: актин(ACT_YEAST), гистон H3 (H3_CAEEL), тубулин(TBB2_CAEEL). Последовательности я получила из RefSeq.
Я использовала Blast+. Первым шагом было создать локальную базу данных. Запрос: makeblastdb -in X5.fasta -dbtype nucl.
Для поиска гомологов я пользовалась tBlastn. Запрос: tblastn -query tubulin.fasta -db X5.fasta -out tubulin.out
Покрытие я посчитала вручную (можно поделить длину лучшего выравнивания на длину белка).
1)Для тубулина получилось: Score = 729 bits (1882), Expect = 0.0, Identities = 371/455 (82%), Positives = 413/455 (91%), Gaps = 22/455 (5%), Frame = -2, Query Cover=96,2%
Судя по значениям,в том числе высокому query cover, можно говорить о гомологии этого белка
2)Для актина получилось: Score = 721 bits (1861), Expect = 0.0, Identities = 333/375 (89%), Positives = 363/375 (97%), Gaps = 0/375 (0%), Frame = -2 , Query cover= 83,3 %.
Можно предположить, что белки гомологичны, но по данным параметрам сходства точно судить мы не можем.
3)Для гистона H3 получилось: Score = 241 bits (615), Expect = 2e-73, Identities = 124/136 (91%), Positives = 127/136 (93%), Gaps = 1/136 (1%), Frame = -3, Query cover= 30%.
В этом случае и по e-value и по query cover можно предположить, что белки негомологичны.
Я взяла скэффолд из организма Amoeboaphelidium protococcarum.
Команда для получения информации о длине контигов: infoseq X5.fasta -only -name -length. Я выбрала scaffold-258 длиной 99209 нуклеотидов(seqret X5.fasta:scaffold-258 -out sc258.out).
Используя blastx, я ограничила поиск царством Fungi (тк мы знаем что организм пренадлежит ему) и выбором находок в refseq.
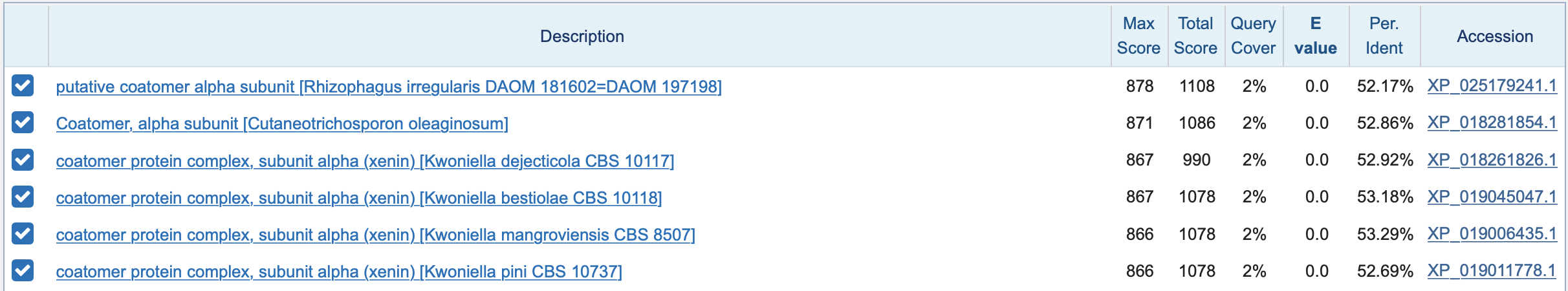
Результаты поиска представлены на картинке. Можно предположить, что ген кодирует coatomer субъединицу альфа.